
NGS library size selection is a critical step in next-generation sequencing (NGS) library preparation that directly impacts data quality, coverage uniformity, and sequencing efficiency. Library size selection isolates DNA fragments within a specific size range, removing both small fragments (including primer/adapter dimers) and larger fragments to boost sequencing efficiency, reduce costs, and improve data quality by ensuring only relevant, correctly sized DNA fragments are sequenced.
Library size selection prevents issues like wasted reads and challenges in alignment. It ensures the sequencer uses its capacity on meaningful data, leading to better assemblies and discoveries.
What is NGS Library Size?
Library size refers to the insert size + adapters (at both ends of the insert):
- Insert size: DNA fragment of interest
- Adapters: platform-specific sequences ligated to both ends of insert fragment
For example: a 300 bp insert with a 70 bp of adapter, the library size will be 300 + 70 + 70 = 440 bp.

What is Library Size Distribution?
NGS Library Size Distribution refers to the range of DNA fragment lengths present in a NGS library after sample preparation. It is a critical quality control metric that directly impacts sequencing efficiency and data quality. An optimal size distribution can get better data by avoiding adapter dimers, increasing cluster efficiency, and maximizing usable reads.
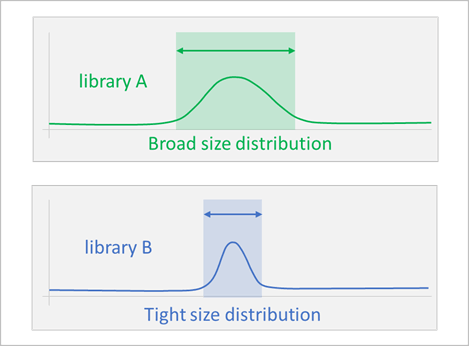
Patterns of library size distribution: Ideal pattern is a tight peak around the target size, rather than a broad, messy spread.
Broad size distributions lead to:
- Uneven amplification
- Coverage bias
- Dropouts in GC-rich or repetitive regions
Tighter size ranges give more consistent coverage, which is critical for:
- Variant calling
- Copy number analysis
- RNA-seq quantification
Factors affecting size distribution:
- Fragmentation method
- Mechanical fragmentation: usually sonication, generates broader distribution
- Enzymatic method: dependent on the type of the enzyme, some are more controlled
- Sample type
- FFPE sample: Often degraded, shorter DNA fragments
- High-quality gDNA: Longer genomic DNA fragments
- RNA-seq: Depends on RNA integrity
Measurement of library size distribution: Gel electrophoresis is used to visualize the size distribution
- Instruments like Bioanalyzer and TapeStation
- Traditional gel electrophoresis
Why Library Size Selection Can Boost Your NGS Results?
Sequencing platforms work best with fragments of certain sizes. Choosing the right fragment length helps:
- Maximizes efficiency: Prevents sequencing capacity from being wasted.
- Uniform fragment sizes: even cluster density
- Reduced phasing/pre-phasing errors
- Higher Q-scores and more stable runs
To maximize sequencing capacity, two approaches can be used:
- Reduce adapter dimers and unwanted short fragments: Adapter dimers are typically ~120-140 bp:
- Amplify extremely efficiently because of the short length
- Can dominate a sequencing run if not removed
- Reduce larger fragments (for short-read sequencing). Large fragments cause low cluster density, and generate less reads and low-quality reads.
- Reduce adapter dimers and unwanted short fragments: Adapter dimers are typically ~120-140 bp:
- Improves data quality: A narrow, uniform fragment size leads to better cluster generation and more accurate read alignment,
- Improves the reliability of genomic assemblies and variant detection (SNPs, indels).
- Reduces Bioinformatics Burden: Cleaner, more uniform libraries mean less time spent filtering out junk reads and dealing with complex data during data analysis.
- Increases reproducibility between samples: This is especially important in clinical, population, and differential expression studies.
When all libraries have similar size profiles:
- Multiplexed samples more evenly
- Reduced batch effects
- More reliable comparisons across experiments
- Optimizes for applications and sequencing platforms:
- Applications: Different applications (whole genome, ChIP-Seq, RNA-seq, long-read) require specific fragment sizes; selection ensures the library matches the specific requirements.
- Sequencing platforms: Different sequencers and sequencing kits have ideal fragment size ranges for efficient cluster generation and sequencing.
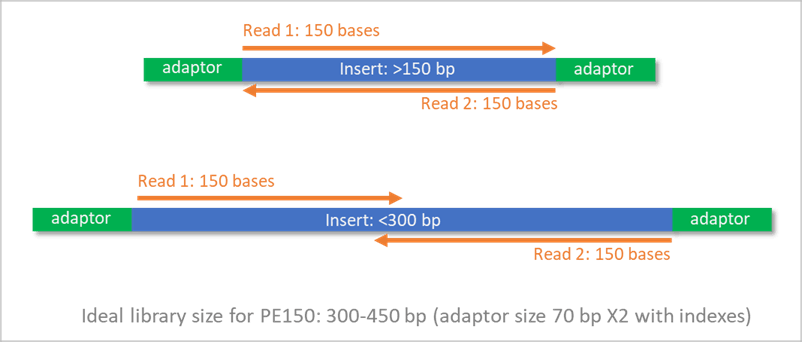
Size selection ensures the insert size fits the sequencing strategy:
- Paired-end reads should overlap minimally or intentionally
- Avoids sequencing adapters instead of DNA inserts
- Optimizes fragment/read alignment
Examples of ideal library sizes:
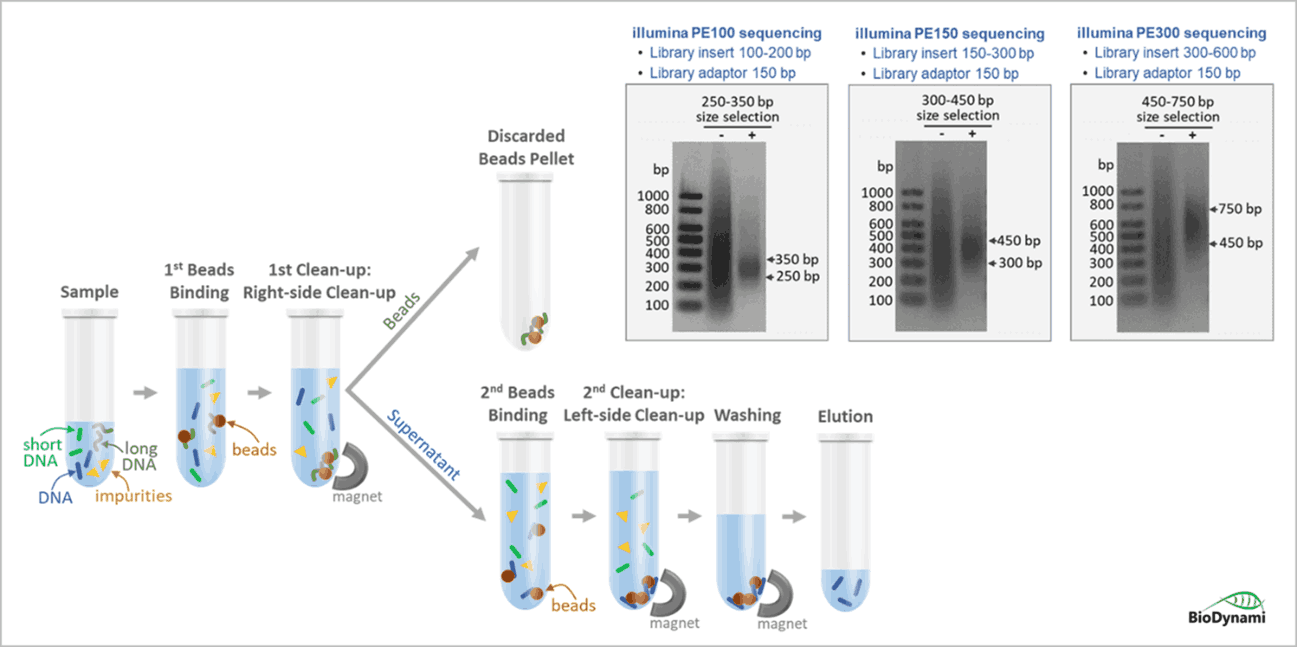
- Illumina PE100 sequencing: ~250–350 bp library size is ideal
- Illumina PE150 sequencing: ~300–450 bp library size is ideal
- Illumina PE300 sequencing: ~450–750 bp library size is ideal
- Long-read sequencing: PacBio, Oxford Nanopore etc., starts from several kb.
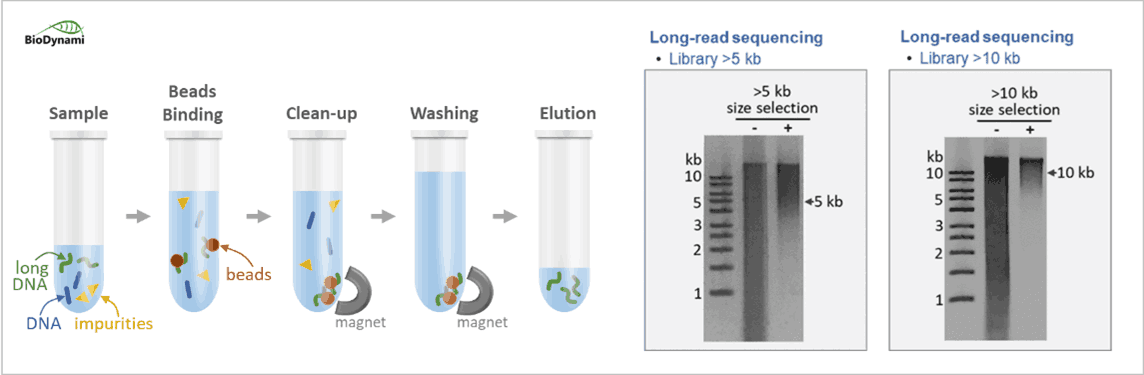
Note: Beads-based kits available for Illumina PE100, PE150, and PE300 with selection of perfect size ranges. For long-read sequencing, >5 kb and >10 kb beads-based size selection kits have been released, indicating beads-based size selection is possible for long DNA fragments.
- Saves money: Reduces sequencing costs by focusing on the most valuable library material.
What Are the Common Issues without Proper Size Selection?
- Too short
- Sequencing adapter-dimers instead of actual library fragments, consume reads but produce no biological information.
- Over-representation of short fragments, leading to inefficient use of sequencing reads
- Too long: Poor cluster formation, low sequencing quality, produce few or low-quality reads
- Broad distribution: Uneven coverage and data loss. Inaccurate assembly and difficult detection of structural variations.
How to Choose the Method for Library Size Selection?
1. Bead-based size selection: This is the most widely adopted method for high-throughput applications.
- Technology: Magnetic beads/SPRI Beads
- Selection type:
- Left-side selection/clean-up: removes small fragments
- Right-side selection/clean-up: removes large fragments
- Double-sided selection/clean-up: selects a tight size range
Example of single-side size selection
Example of double-side size selection
- Mechanism: It uses the principle of solid phase reversible immobilization (SPRI), where the ratio of PEG/salt/beads to DNA determines which fragment sizes bind to the beads. The beads method simplifies workflows by eliminating gels/spin columns and is adaptable for both single-sided and double-sided selections.
- Pros: Highly scalable, reproducible, cost-effective, easily automated, and ideal for removing small fragments like adapter-dimers.
- Automation & High-Throughput: Magnetic beads are ideal for automated liquid handlers, reducing manual pipetting and enabling processing of many samples efficiently.
- Reproducibility: Automation leads to less variability and more consistent results across experiments.
- Cost-Effectiveness: Reagent costs are often lower than other methods, and avoiding spin columns reduces overall costs.
- Efficient Cleanup: Removes both small fragments (primer dimers, adapter dimers) and large fragments, enhances data quality, and maximizes usable reads.
- Simplicity: Straightforward simple protocols.
- Cons:
2. Gel-based size selection: This approach physically separates fragments by electrophoresis.
- Selection type
- Agarose gel electrophoresis: Manual but highly customizable; allows precise excision of desired fragment sizes.
- Precise but labor-intensive and lower yield
- Often used for special applications (e.g., mate-pair libraries)
- Pippin Prep/BluePippin (Sage Science): Automated gel-based systems. Recent data shows these remain popular for challenging applications like ATAC-seq and mate-pair libraries.
- Agarose gel electrophoresis: Manual but highly customizable; allows precise excision of desired fragment sizes.
- Mechanism: DNA runs through an agarose gel, and the desired size range is physically extracted or electroeluted.
- Pros:
- Highly accurate for precise size ranges
- reproducible
- Cons:
- Low throughput. Automated gel systems like Pippin Prep address some throughput issues but still have limitations compared to bead-based methods.
- Labor-intensive and time-consuming.
- Potential for contamination
- DNA loss (typically 30-50%)
